Research
Electronic structure & reactivity
I use electronic-structure theory — density functional theory, and methods beyond it — in close collaboration with experimentalists, to explain how metals, clusters and functional molecules bind and transform. The unifying question is how the immediate environment of a metal or a cluster controls its oxidation state, spin, reactivity and the routing of electrons. Several linked themes run through the work.
Cobalamin (vitamin B12) reactivity
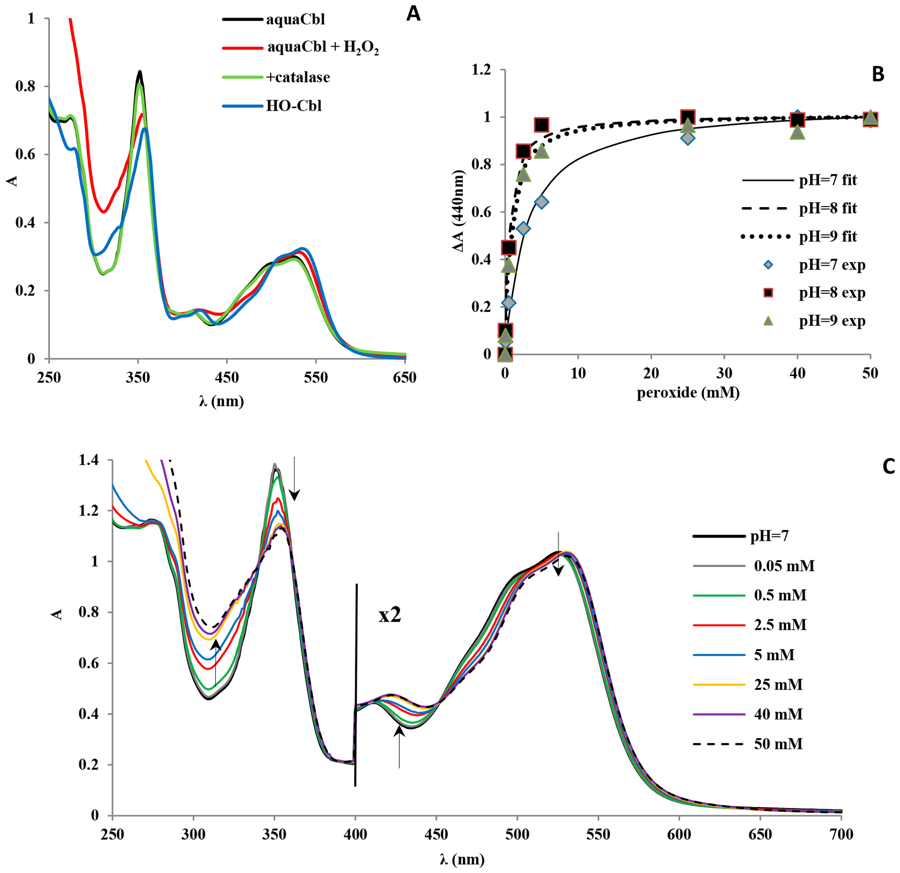
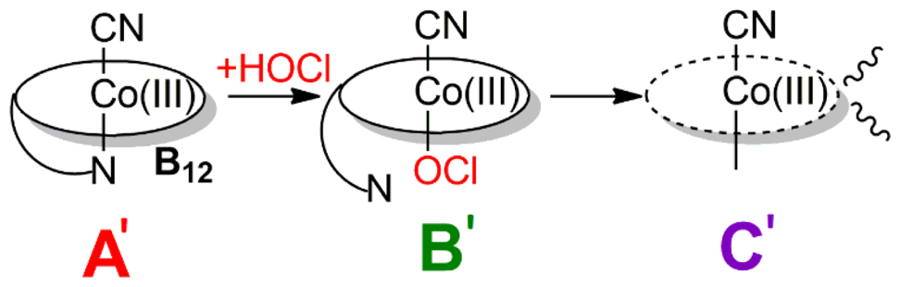
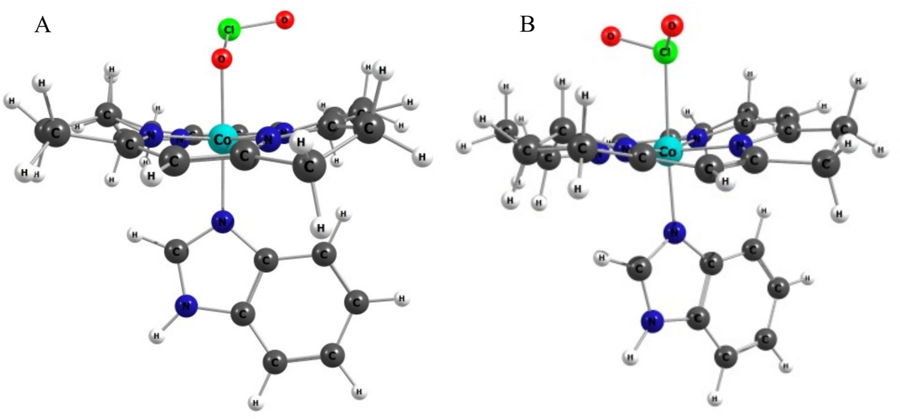
The cobalt corrin at the heart of vitamin B12 is a remarkably versatile redox and coordination platform. In a long-running collaboration I have helped show that its Co(III) centre forms a whole family of axial adducts once thought out of reach — with hydrogen peroxide[10], organic peroxides[17], hypochlorite[16], chlorite[25] and oxidised-cysteine derivatives[18] — each pinned down by matching experiment to DFT and TD-DFT. One of them, a cobalt–chlorite complex, turns out to be a stable analogue of the first intermediate in the catalytic cycle of the heme enzyme chlorite dismutase[33].
Heme, siroheme & the logic of the macrocycle
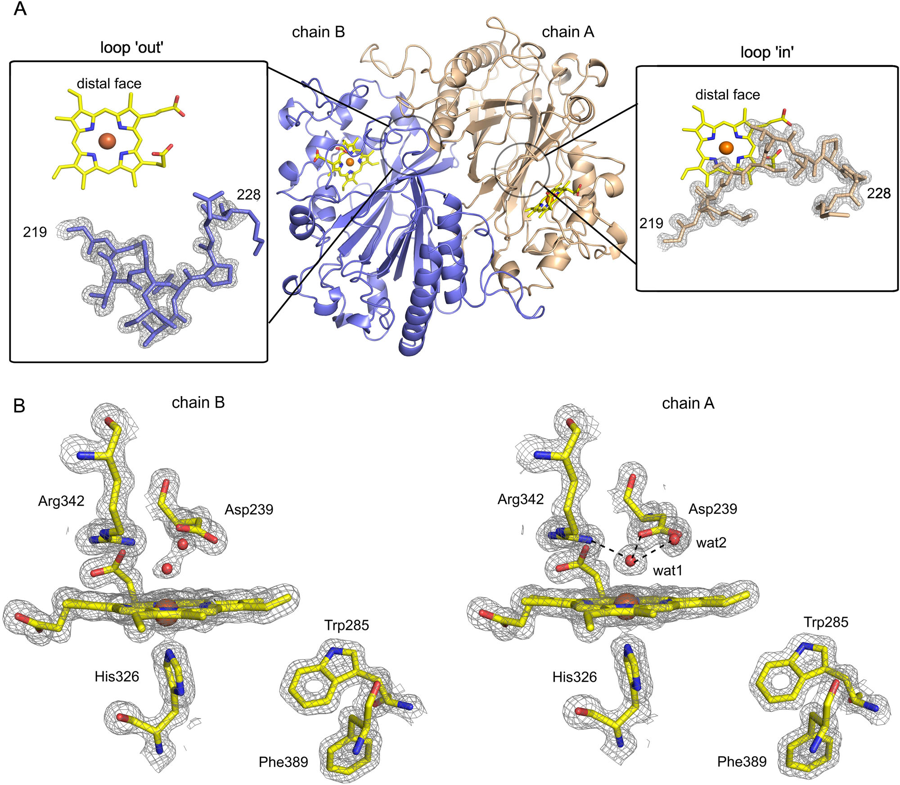
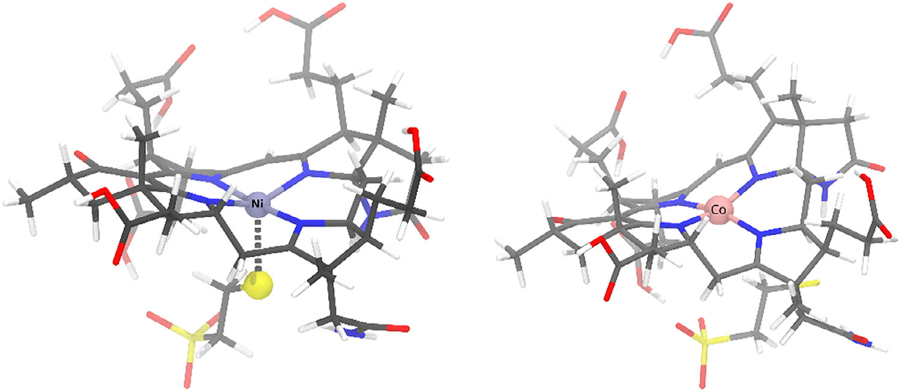
The same iron behaves very differently inside heme, siroheme or a corrin, and much of my work asks how the macrocycle sets the chemistry. Using electron-transport (Green’s-function) methods I explained why sulfite reductase employs siroheme rather than ordinary heme[6]: the modified ring suppresses stray charge-transfer routes and channels electrons cleanly to the catalytic iron[8]. Earlier work set out the ferrous–sulfite (FeSO2) intermediates of the enzyme’s own reaction[5]. I also study high-valent ferryl intermediates[26] — contributing the computational analysis to a 2026 Nature Communications study showing that ferryl species in a heme peroxidase owe their unexpectedly long Fe–O bonds to accessible ferric-oxyl excited states[31]. A related study rationalises why biology pairs cobalt with the corrin of B12 but nickel with the corphin of coenzyme F430, tracing it to the conjugation of the ring[32].
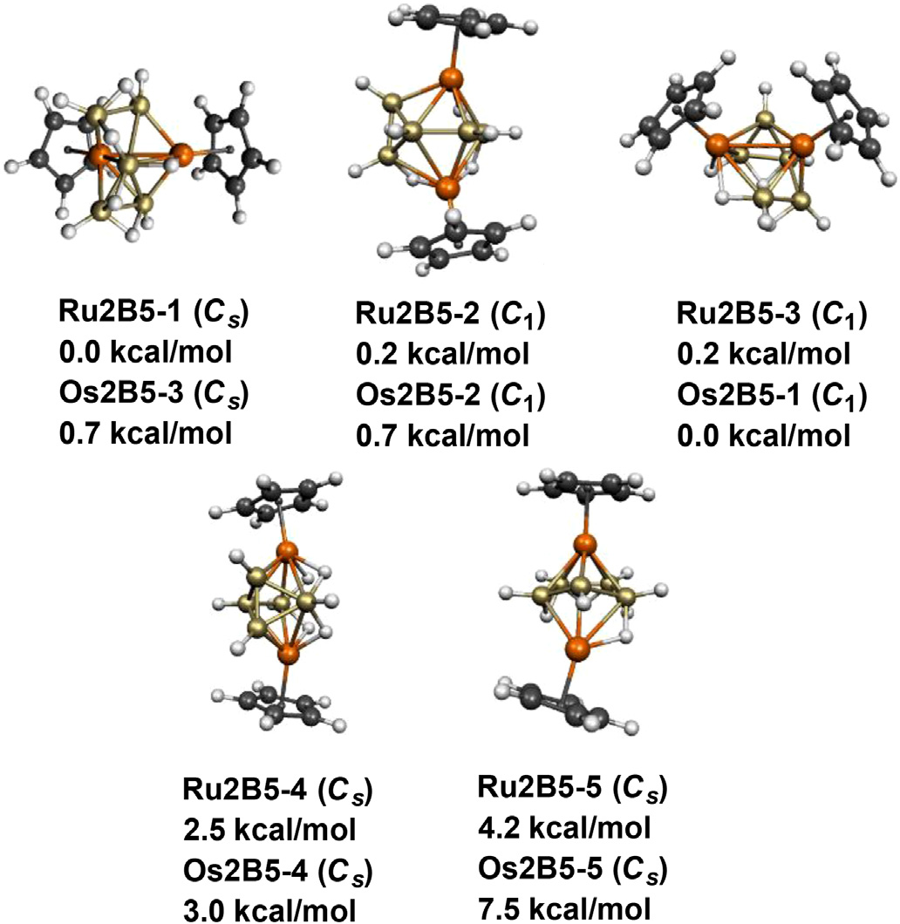
Cluster & main-group bonding
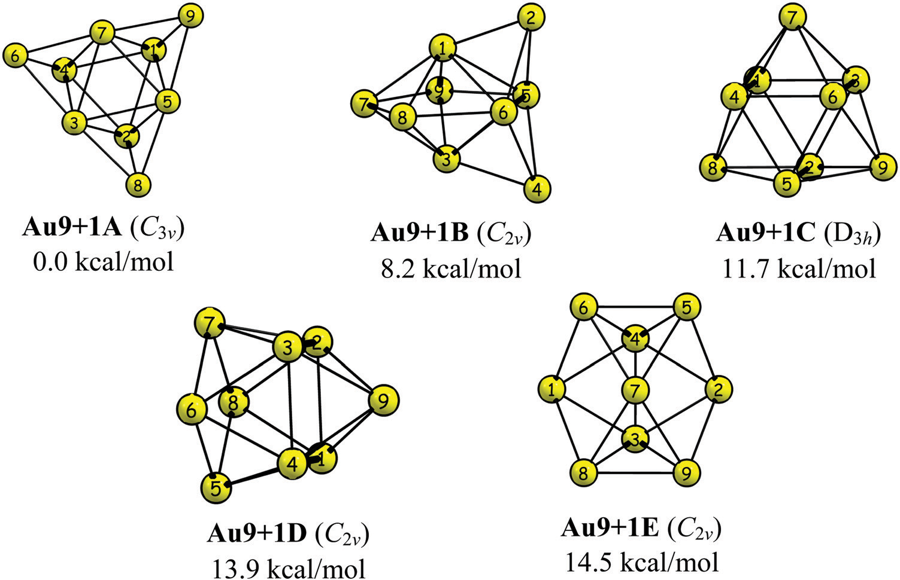
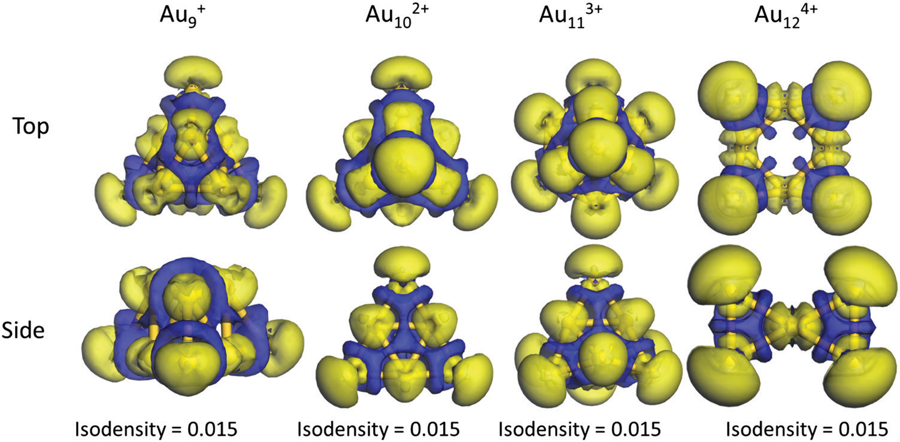
With Alexandru Lupan and R. Bruce King I study how the number of skeletal electrons dictates the shape of electron-deficient cages. A series of studies mapped how hydrogen-rich dimetallaboranes obey the Wade–Mingos electron-counting rules — reproducing and rationalising the experimentally known structures[1, 2, 3, 4] — and how cationic gold “superatom” clusters display spherical aromaticity and surface σ-holes[7], connecting the language of borane chemistry to bare-metal clusters.
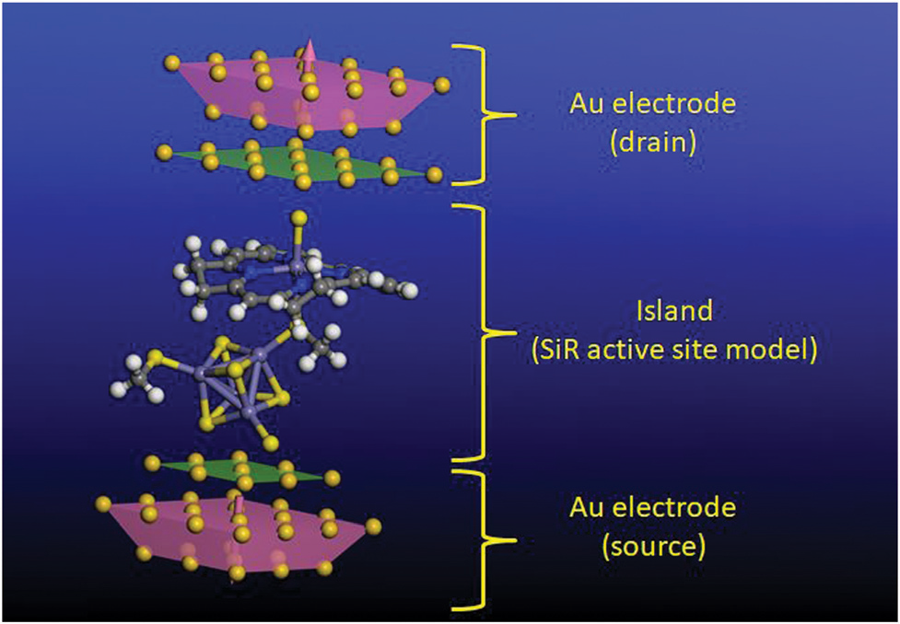
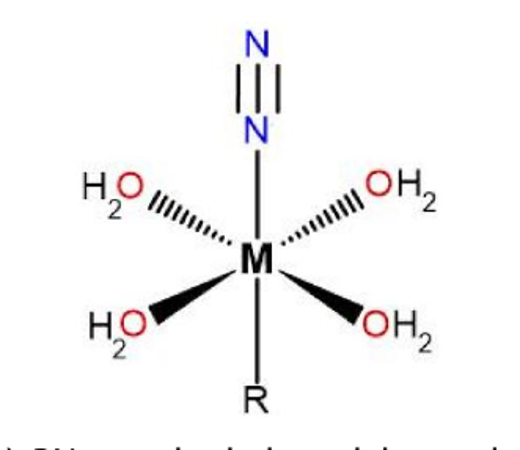
Binding & activating small molecules
Whether a metal can grip an inert molecule like dinitrogen and make it react depends sensitively on its coordination environment. I study how the ligand field around iron[19] — and around manganese and cobalt — tunes the binding and activation of N2, finding that low oxidation states and carbanion ligands are decisive[23]. The work bears directly on the nitrogenase mechanism and on the question of whether an “iron-free” nitrogenase is feasible.
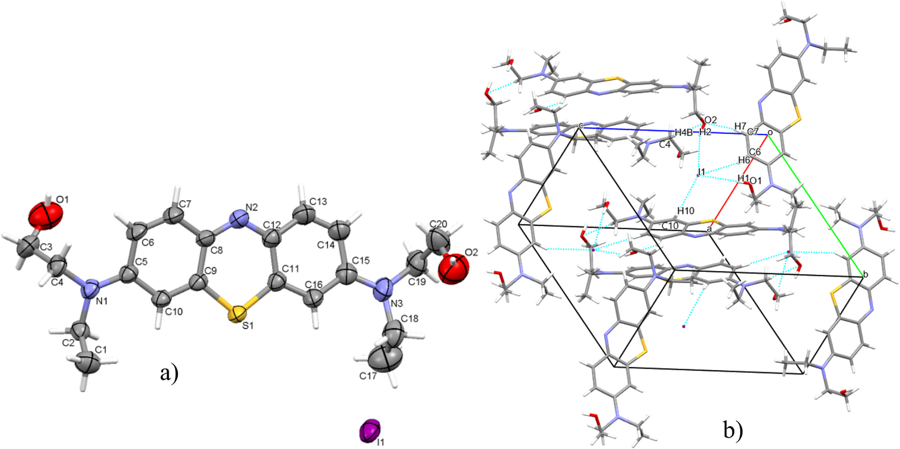
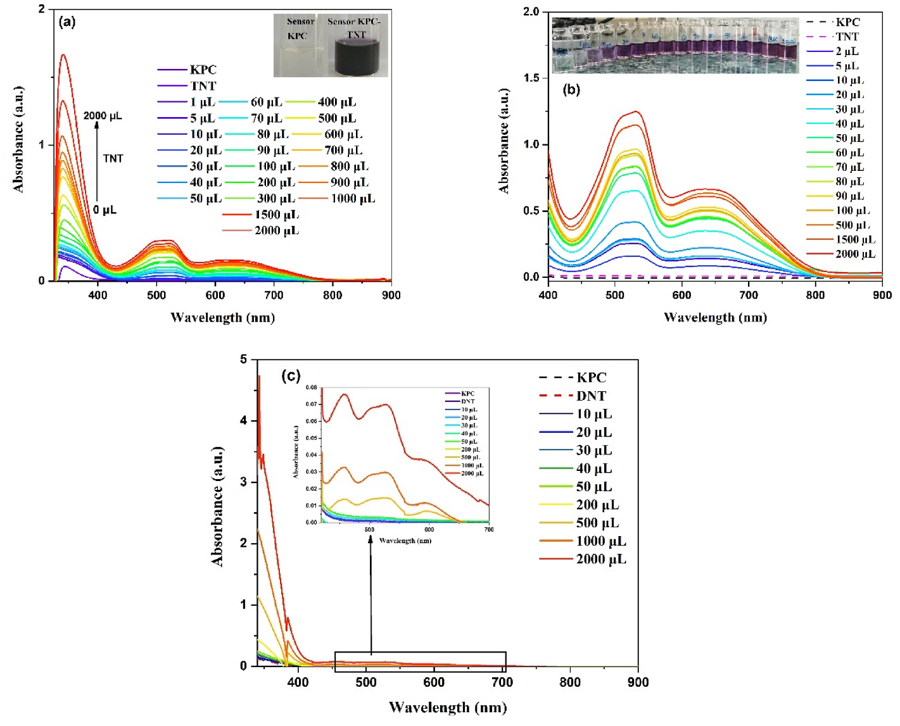
Photofunctional molecules & applied computation
I provide the excited-state and electronic-structure analysis for functional molecules and materials developed with experimental groups in Cluj-Napoca. This includes phenothiazine and phenothiazinium dyes for fluorescence imaging[21], photodynamic therapy (as singlet-oxygen photosensitisers)[15, 27] and the colorimetric sensing of nitroaromatic explosives[34], together with computational support for green corrosion inhibitors[22, 29, 30] and for luminescent zinc[24] and europium[28] materials.
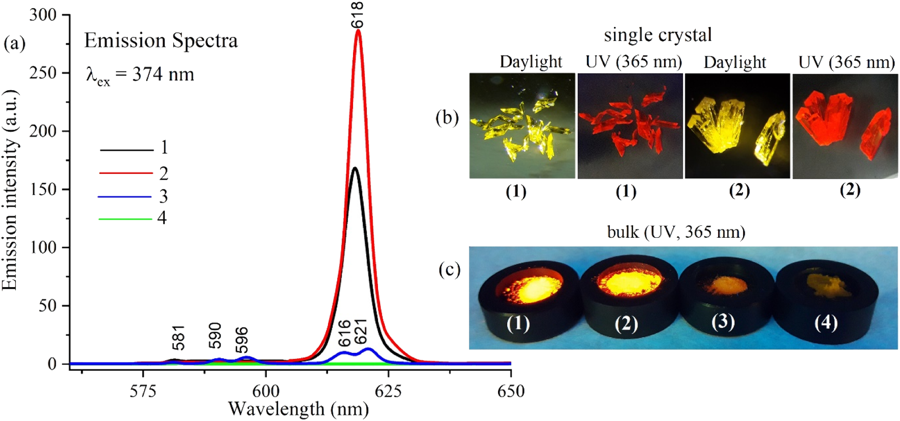
Spectroscopy, redox & structure
Computed electronic structure is most powerful when anchored to experiment. I use it to interpret and predict spectra — including a fresh look at one of chemistry’s most familiar demonstrations, the intense blue of the iodine–starch complex[14] — and to unpick reaction mechanisms, such as the selenium and sulfur redox chemistry that generates reactive radicals in biology[9, 11]. The same toolkit of classical and QM/MM molecular dynamics follows larger assemblies in motion, from the non-heme-iron protein hemerythrin[13] to polylactic acid at a bioceramic surface[12].
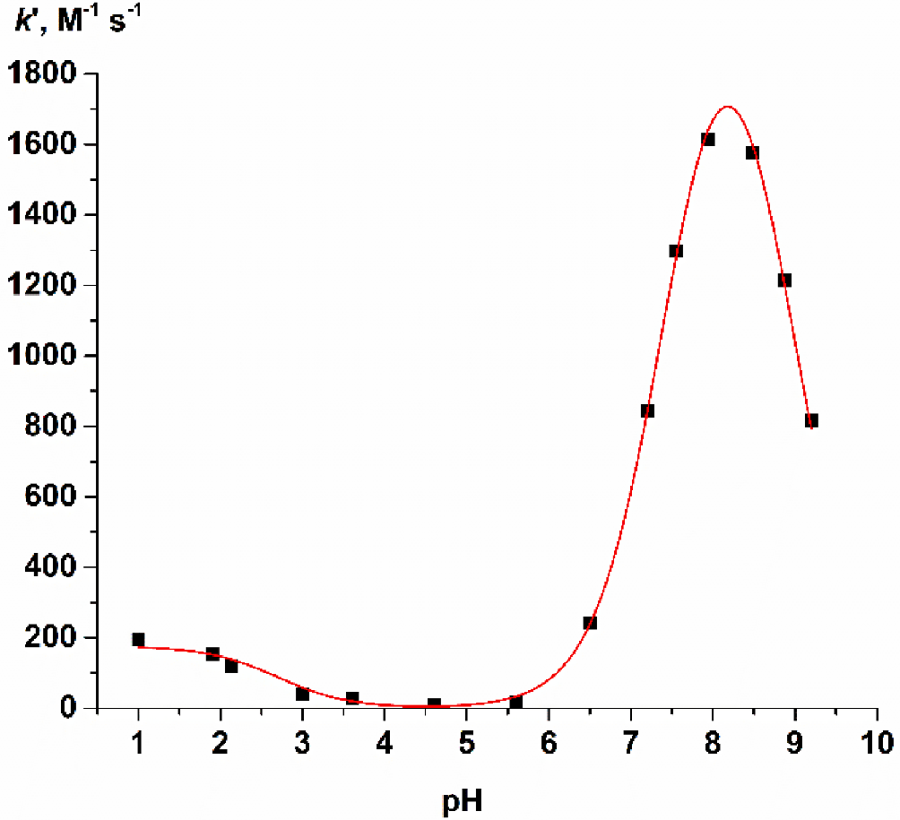
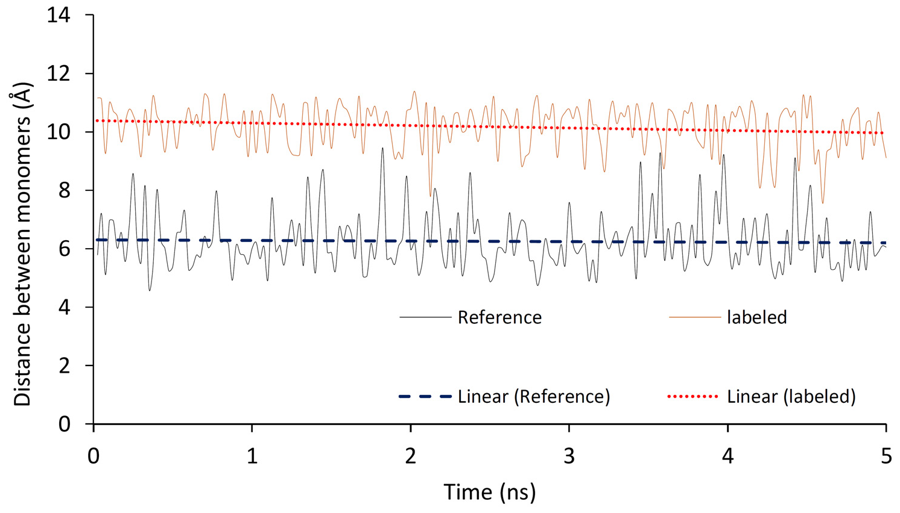
Foundations of quantum chemistry
Alongside the calculations I keep a distinct interest in the foundations of the theory itself — a project I call Quale Mechanics, a reading of the qualitative content of quantum mechanics[20], and an account of the covalent bond as a stabilised “Fermi heap”. The aim is the same as in the computational work: to say precisely what is happening beneath a familiar chemical picture, and to map uncertainty openly rather than hide it.
See the underlying papers
Each theme is backed by peer-reviewed work with collaborators in Cluj-Napoca and beyond — thirty-four articles from 2014 to 2026.
Browse publications